Model Fitting
model_fitting.RmdHere we describe the basic steps of joint model fitting with
{jmpost}.
Model Specification
Each JointModel needs to be specified with three
parts:
-
longitudinal: The model for the longitudinal outcomes. -
survival: The model for the survival outcomes. -
link: The link that specifies how thelongitudinalmodel parameters enter thesurvivalmodel.
Default Options
Let’s first specify a very simple joint model with:
- A random slope model for the longitudinal outcome.
- A Weibull proportional hazards model for the survival outcome.
- The link between the two models is that the random slope from the longitudinal model enters as a product with a link coefficient into the linear predictor of the survival model.
simple_model <- JointModel(
longitudinal = LongitudinalRandomSlope(),
survival = SurvivalWeibullPH(),
link = linkDSLD()
)Note that here we use all the default options for the two models and the link, in particular the prior distributions and the initial values in the MCMC chain for the parameters are automatically chosen. We can see this from the arguments of the constructors (or from the help page):
args(LongitudinalRandomSlope)
#> function (intercept = prior_normal(30, 10), slope_mu = prior_normal(1,
#> 3), slope_sigma = prior_lognormal(0, 1.5), sigma = prior_lognormal(0,
#> 1.5))
#> NULLSo here we see that the Longitudinal Random Slope model has 4 parameters that we can define a prior for.
Specifying Priors
We can alternatively also specify the prior distributions for the parameters manually. This is important in practice to obtain a meaningful model specification and hence converging MCMC chains that allow to estimate the posterior distributions.
For the random slope model for the longitudinal outcome, we can e.g. say:
random_slope_model <- LongitudinalRandomSlope(
intercept = prior_normal(40, 5),
slope_mu = prior_normal(10, 2)
)This sets the prior for the intercept to be a \(N(40, 5)\) distribution and the prior for
the slope_mu parameter to be a \(N(10, 2)\) distribution.
Separate Models
It is also possible to not link the longitudinal and the survival
models, by using the special linkNone() link specification.
For example,
simple_model_no_link <- JointModel(
longitudinal = LongitudinalRandomSlope(),
survival = SurvivalWeibullPH(),
link = linkNone()
)would allow to fit the two models separately, but in the same MCMC chain.
Single Models
It is also possible to specify only the longitudinal or the survival model. Then these can be estimated on their own with separate MCMC chains.
single_longitudinal <- JointModel(
longitudinal = LongitudinalRandomSlope()
)
single_survival <- JointModel(
survival = SurvivalWeibullPH()
)Data Preparation
Before we can fit the models, we need to prepare the data in the right format.
Simulating Data
Here we start from a simulated data set.
- We assign 50 subjects each to the two treatment arms.
- We use a time grid from 1 to 2000, e.g. specifying the days after randomization.
- We use an exponentially distributed censoring time with mean of 9000 days.
- We use a categorical covariate with three levels A, B and C in the overall survival model, drawn uniformly from the three levels. (Note that this is hardcoded at the moment, so the levels need to be A, B, C.)
- We use another continuous covariate in the overall survival model generated from a standard normal distribution, with coefficient 0.3.
- For the longitudinal outcome, we draw the values from a random slope model with the given parameters.
- For the survival outcome, we draw the true value from a Weibull
model. Note that it is fairly easy to put here another choice, you just
need to specify a function of
timereturning the log baseline hazard under the given survival model.
So let’s run the code for that:
set.seed(129)
sim_data <- SimJointData(
design = list(
SimGroup(50, "Arm-A", "Study-X"),
SimGroup(50, "Arm-B", "Study-X")
),
longitudinal = SimLongitudinalRandomSlope(
times = c(1, 50, 100, 150, 200, 250, 300),
intercept = 30,
slope_mu = c(1, 2),
slope_sigma = 0.2,
sigma = 20,
link_dsld = 0.1
),
survival = SimSurvivalWeibullPH(
lambda = 1 / 300,
gamma = 0.97,
time_max = 2000,
time_step = 1,
lambda_cen = 1 / 9000,
beta_cat = c(
"A" = 0,
"B" = -0.1,
"C" = 0.5
),
beta_cont = 0.3
)
)We might get a message here that a few subjects did not die before the day 2000, but this is not of concern. Basically it just gives us a feeling of how many survival events are included in the data set.
Formatting Data
Next we bring the data into the right format.
We start with extracting data into individual data sets, and then reducing the longitudinal data to specific time points.
os_data <- sim_data@survival
long_data <- sim_data@longitudinalLet’s have a quick look at the format:
The survival data has:
- subject ID
- time point
- continuous covariate value
- categorical covariate level
- event indicator (1 for observed, 0 for censored)
- study ID
- treatment arm
head(os_data)
#> # A tibble: 6 × 7
#> subject study arm time event cov_cont cov_cat
#> <chr> <fct> <fct> <dbl> <dbl> <dbl> <fct>
#> 1 subject_001 Study-X Arm-A 35 1 -1.12 B
#> 2 subject_002 Study-X Arm-A 17 1 -0.990 C
#> 3 subject_003 Study-X Arm-A 876 1 -1.37 C
#> 4 subject_004 Study-X Arm-A 100 1 -1.36 C
#> 5 subject_005 Study-X Arm-A 9 1 2.00 B
#> 6 subject_006 Study-X Arm-A 122 1 0.696 BThe longitudinal data has:
- subject ID
- time point
- sum of longest diameters (SLD)
- study ID
- treatment arm
- observation flag
head(long_data)
#> # A tibble: 6 × 6
#> subject arm study time sld observed
#> <chr> <fct> <fct> <dbl> <dbl> <lgl>
#> 1 subject_001 Arm-A Study-X 1 23.4 TRUE
#> 2 subject_001 Arm-A Study-X 50 108. FALSE
#> 3 subject_001 Arm-A Study-X 100 137. FALSE
#> 4 subject_001 Arm-A Study-X 150 141. FALSE
#> 5 subject_001 Arm-A Study-X 200 220. FALSE
#> 6 subject_001 Arm-A Study-X 250 325. FALSEFinally, we wrap these in the data formatting functions. Here the mapping of the column names to the required variables happens. This means that in our applications we don’t have to use the same variable names as seen above, but we can use custom names and then apply the mapping here.
joint_data <- DataJoint(
subject = DataSubject(
data = os_data,
subject = "subject",
arm = "arm",
study = "study"
),
survival = DataSurvival(
data = os_data,
formula = Surv(time, event) ~ cov_cat + cov_cont
),
longitudinal = DataLongitudinal(
data = long_data,
formula = sld ~ time,
threshold = 5
)
)Model Fitting
Now let’s have a look how we can fit the (joint) models.
Debugging Stan Code
It is always possible to read out the Stan code that is contained in
the JointModel object, using write_stan():
tmp <- tempfile()
write_stan(simple_model, destination = tmp)
first_part <- head(readLines(tmp), 10)
cat(paste(first_part, collapse = "\n"))
#> functions {
#> //
#> // Source - base/functions.stan
#> //
#>
#> // Constant used in below.
#> real neg_log_sqrt_2_pi() {
#> return -0.9189385332046727;
#> }Sampling Parameters
Finally, sampleStanModel() is kicking off the MCMC
sampler via cmdstanr in the backend. Note that in practice
you would increase the number of warm-up and sampling iterations.
mcmc_results <- sampleStanModel(
simple_model,
data = joint_data,
iter_sampling = 500,
iter_warmup = 500,
chains = 1,
parallel_chains = 1
)
#> Running MCMC with 1 chain...
#>
#> Chain 1 Iteration: 1 / 1000 [ 0%] (Warmup)
#> Chain 1 Informational Message: The current Metropolis proposal is about to be rejected because of the following issue:
#> Chain 1 Exception: gamma_lpdf: Random variable is inf, but must be positive finite! (in '/tmp/Rtmpb6WNy1/model-19a1625d1533.stan', line 507, column 4 to column 100)
#> Chain 1 If this warning occurs sporadically, such as for highly constrained variable types like covariance matrices, then the sampler is fine,
#> Chain 1 but if this warning occurs often then your model may be either severely ill-conditioned or misspecified.
#> Chain 1
#> Chain 1 Iteration: 100 / 1000 [ 10%] (Warmup)
#> Chain 1 Iteration: 200 / 1000 [ 20%] (Warmup)
#> Chain 1 Iteration: 300 / 1000 [ 30%] (Warmup)
#> Chain 1 Iteration: 400 / 1000 [ 40%] (Warmup)
#> Chain 1 Iteration: 500 / 1000 [ 50%] (Warmup)
#> Chain 1 Iteration: 501 / 1000 [ 50%] (Sampling)
#> Chain 1 Iteration: 600 / 1000 [ 60%] (Sampling)
#> Chain 1 Iteration: 700 / 1000 [ 70%] (Sampling)
#> Chain 1 Iteration: 800 / 1000 [ 80%] (Sampling)
#> Chain 1 Iteration: 900 / 1000 [ 90%] (Sampling)
#> Chain 1 Iteration: 1000 / 1000 [100%] (Sampling)
#> Chain 1 finished in 24.9 seconds.Convergence checks
After the sampling finishes, we can inspect the parameter
distributions. This is using the cmdstanr functions,
because the results element is of class
CmdStanMCMC.
vars <- c(
"lm_rs_intercept",
"lm_rs_slope_mu",
"lm_rs_slope_sigma",
"lm_rs_sigma",
"link_dsld",
"sm_weibull_ph_lambda",
"sm_weibull_ph_gamma",
"beta_os_cov"
)
as.CmdStanMCMC(mcmc_results)$summary(vars)
#> # A tibble: 11 × 10
#> variable mean median sd mad q5 q95 rhat ess_bulk
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 lm_rs_inte… 2.77e+1 27.7 1.29 1.31 25.4 29.7 1.00 254.
#> 2 lm_rs_slop… 1.02e+0 1.02 0.0296 0.0285 0.972 1.07 0.998 775.
#> 3 lm_rs_slop… 1.96e+0 1.96 0.0296 0.0303 1.91 2.01 1.00 938.
#> 4 lm_rs_slop… 2.04e-1 0.203 0.0148 0.0158 0.181 0.229 1.00 938.
#> 5 lm_rs_sigma 1.92e+1 19.2 0.543 0.570 18.4 20.1 0.999 799.
#> 6 link_dsld -5.73e-2 -0.0472 0.200 0.188 -0.391 0.277 1.00 539.
#> 7 sm_weibull… 1.00e-2 0.00863 0.00562 0.00440 0.00371 0.0204 1.00 372.
#> 8 sm_weibull… 8.85e-1 0.882 0.0711 0.0755 0.770 1.00 1.00 689.
#> 9 beta_os_co… -1.53e-5 -0.00640 0.239 0.233 -0.372 0.399 1.00 764.
#> 10 beta_os_co… 3.34e-1 0.333 0.261 0.264 -0.0937 0.750 1.00 866.
#> 11 beta_os_co… 3.65e-1 0.370 0.104 0.0972 0.190 0.531 1.00 1167.
#> # ℹ 1 more variable: ess_tail <dbl>We can already see here from the rhat statistic whether
the MCMC sampler converged - values close to 1 indicate convergence,
while values larger than 1 indicate divergence.
In general, convergence is sensitive to the choice of:
- Priors
- Initial values
- Sufficient warm-up iterations
If you don’t achieve convergence, then play around with different choices of the above.
Plotting
We can now proceed towards investigating the results of the MCMC chain with plots.
Trace plots
Using the draws() method on the results
element, we can extract the samples in a format that is understood by
bayesplot. We can e.g. look at some simple trace plots:
vars_draws <- as.CmdStanMCMC(mcmc_results)$draws(vars)
library(bayesplot)
#> This is bayesplot version 1.11.1
#> - Online documentation and vignettes at mc-stan.org/bayesplot
#> - bayesplot theme set to bayesplot::theme_default()
#> * Does _not_ affect other ggplot2 plots
#> * See ?bayesplot_theme_set for details on theme setting
mcmc_trace(vars_draws)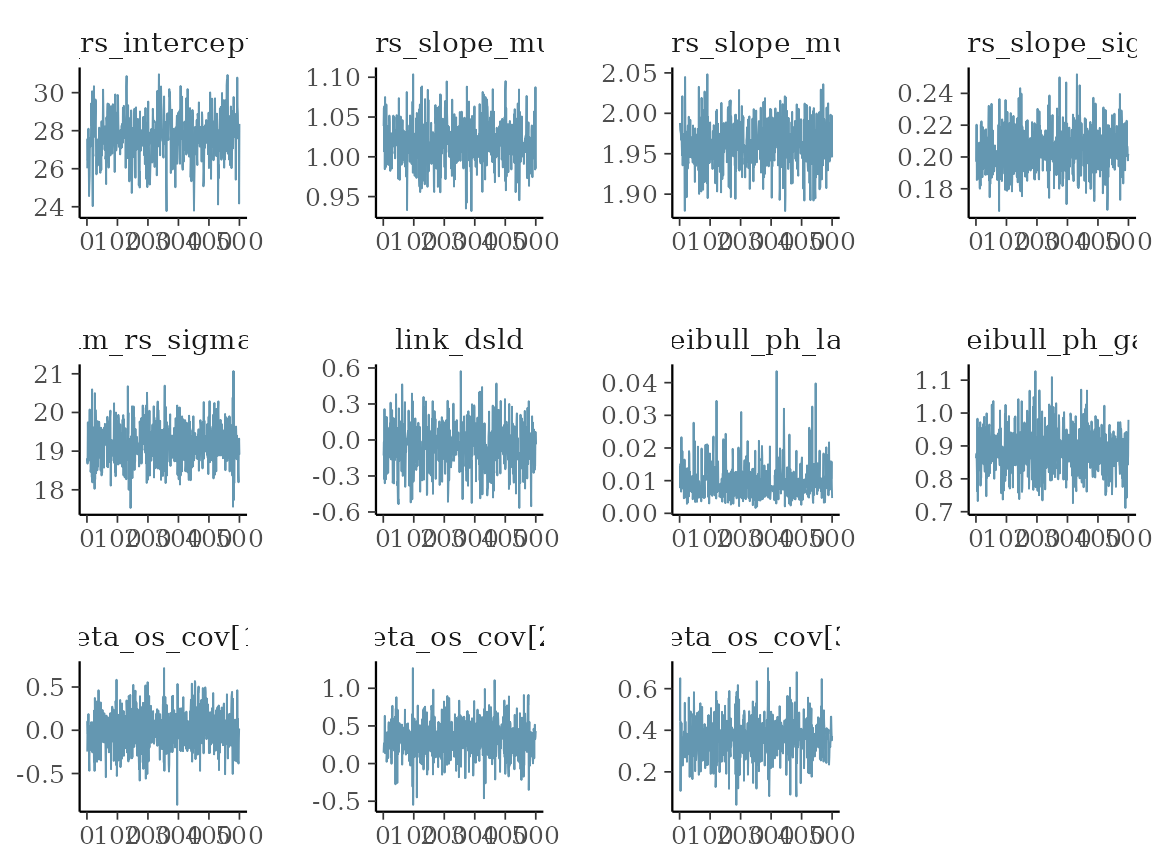
Longitudinal fit plots
Using the longitudinal() method we can extract the
longitudinal fit samples from the result, and then plot them for all
subjects or those that we are interested in. For illustration, we will
plot here the first 10 subjects:
selected_subjects <- head(os_data$subject, 10)
long_quantities <- LongitudinalQuantities(
mcmc_results,
grid = GridFixed(
subjects = selected_subjects
)
)
as.data.frame(long_quantities) |> head()
#> group time values
#> 1 subject_001 0 26.0238
#> 2 subject_001 0 27.5690
#> 3 subject_001 0 26.4398
#> 4 subject_001 0 26.6774
#> 5 subject_001 0 28.1050
#> 6 subject_001 0 26.0869
summary(long_quantities) |> head()
#> group time median lower upper
#> 1 subject_001 0 27.69405 25.14073 30.12683
#> 2 subject_002 0 27.69405 25.14073 30.12683
#> 3 subject_003 0 27.69405 25.14073 30.12683
#> 4 subject_004 0 27.69405 25.14073 30.12683
#> 5 subject_005 0 27.69405 25.14073 30.12683
#> 6 subject_006 0 27.69405 25.14073 30.12683
autoplot(long_quantities)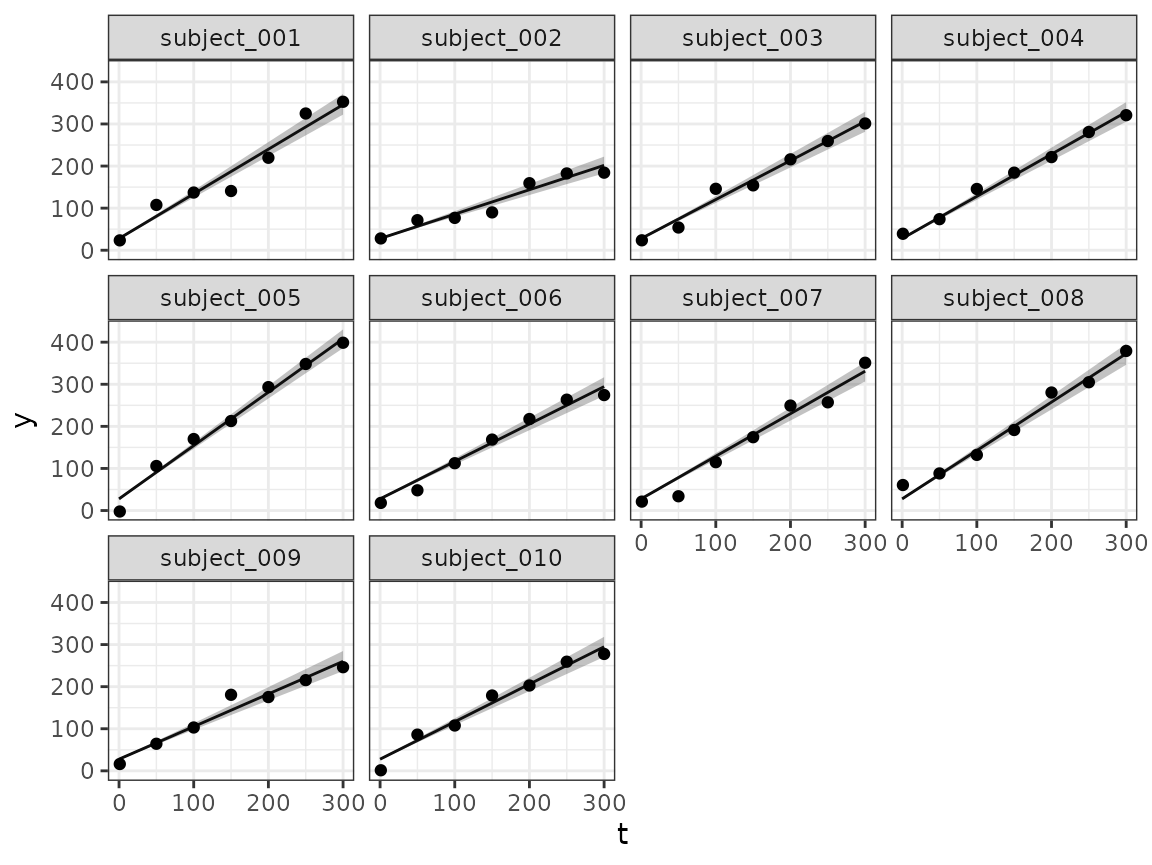
Survival fit plots
And using the SurvivalQuantities() method we can do the
same for the estimated survival functions.
surv_quantities <- SurvivalQuantities(
mcmc_results,
grid = GridFixed(
subjects = selected_subjects
)
)
as.data.frame(surv_quantities) |> head()
#> group time values
#> 1 subject_001 0 1
#> 2 subject_001 0 1
#> 3 subject_001 0 1
#> 4 subject_001 0 1
#> 5 subject_001 0 1
#> 6 subject_001 0 1
summary(surv_quantities) |> head()
#> group time median lower upper
#> 1 subject_001 0 1 1 1
#> 2 subject_002 0 1 1 1
#> 3 subject_003 0 1 1 1
#> 4 subject_004 0 1 1 1
#> 5 subject_005 0 1 1 1
#> 6 subject_006 0 1 1 1
autoplot(surv_quantities)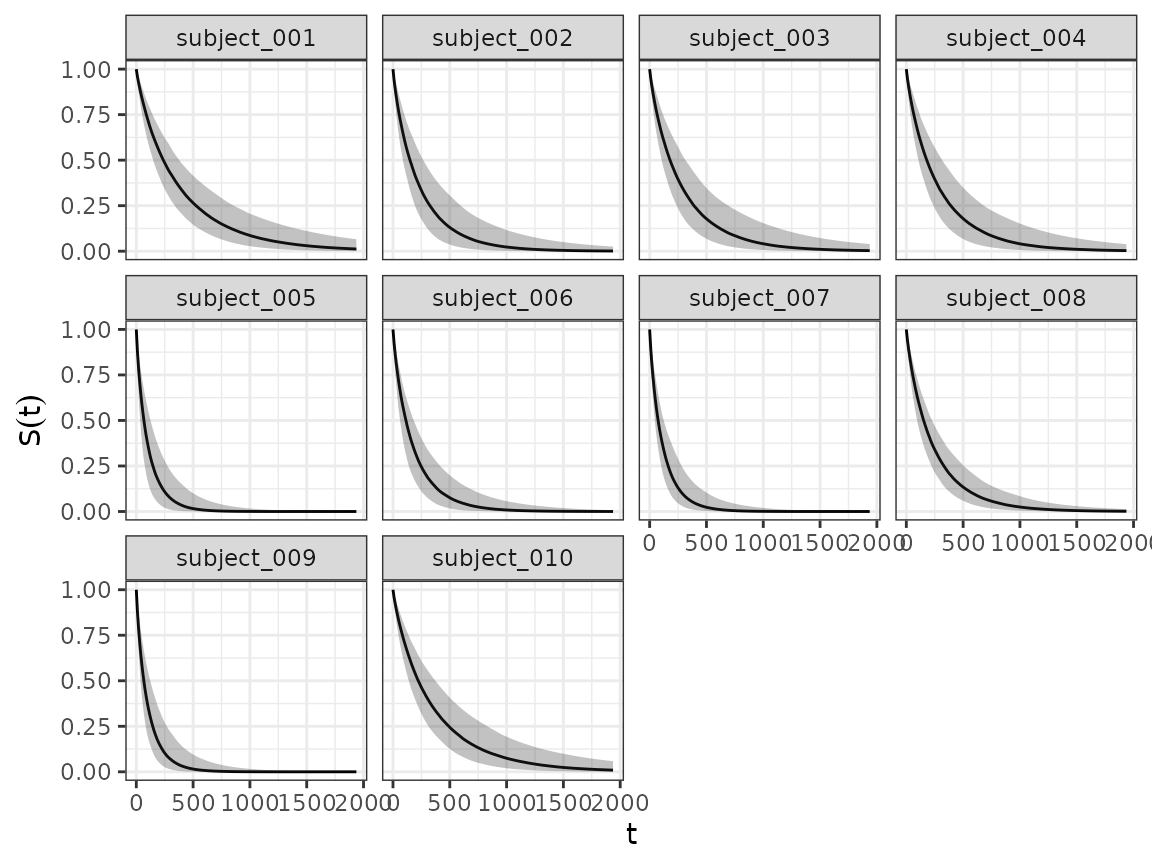
We can also aggregate the estimated survival curves from groups of subjects, using the corresponding method.
surv_quantities <- SurvivalQuantities(
mcmc_results,
grid = GridGrouped(
groups = split(os_data$subject, os_data$arm)
)
)
autoplot(surv_quantities, add_km = TRUE)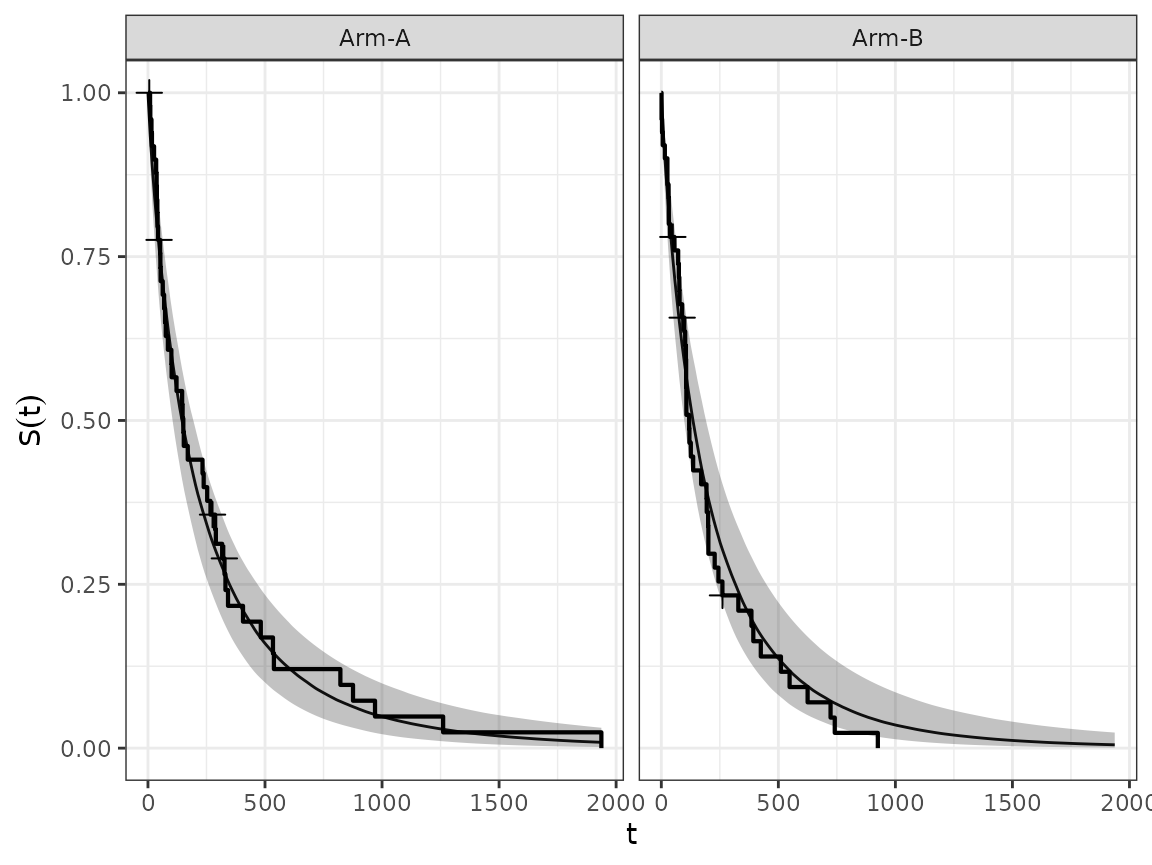
Predicting Survival Quantities for Hypothetical Subjects
The SurvivalQuantities() method can also be used to
predict survival quantities for hypothetical subjects with any arbitrary
covariate and longitudinal model parameter values. This is done via
passing a GridPrediction() object to the method where the
newdata and params arguments are used to
specify the desired values for the covariates and longitudinal model
parameters respectively. For example below we use these functions to
predict the survival distribution for a hypothetical subject:
surv_quantities <- SurvivalQuantities(
mcmc_results,
grid = GridPrediction(
times = seq(1, 800, by = 50),
newdata = dplyr::tibble(
cov_cat = c("A", "B"),
cov_cont = c(1.2, 2)
),
params = list(
intercept = 40,
slope = 0.05
)
)
)
autoplot(surv_quantities)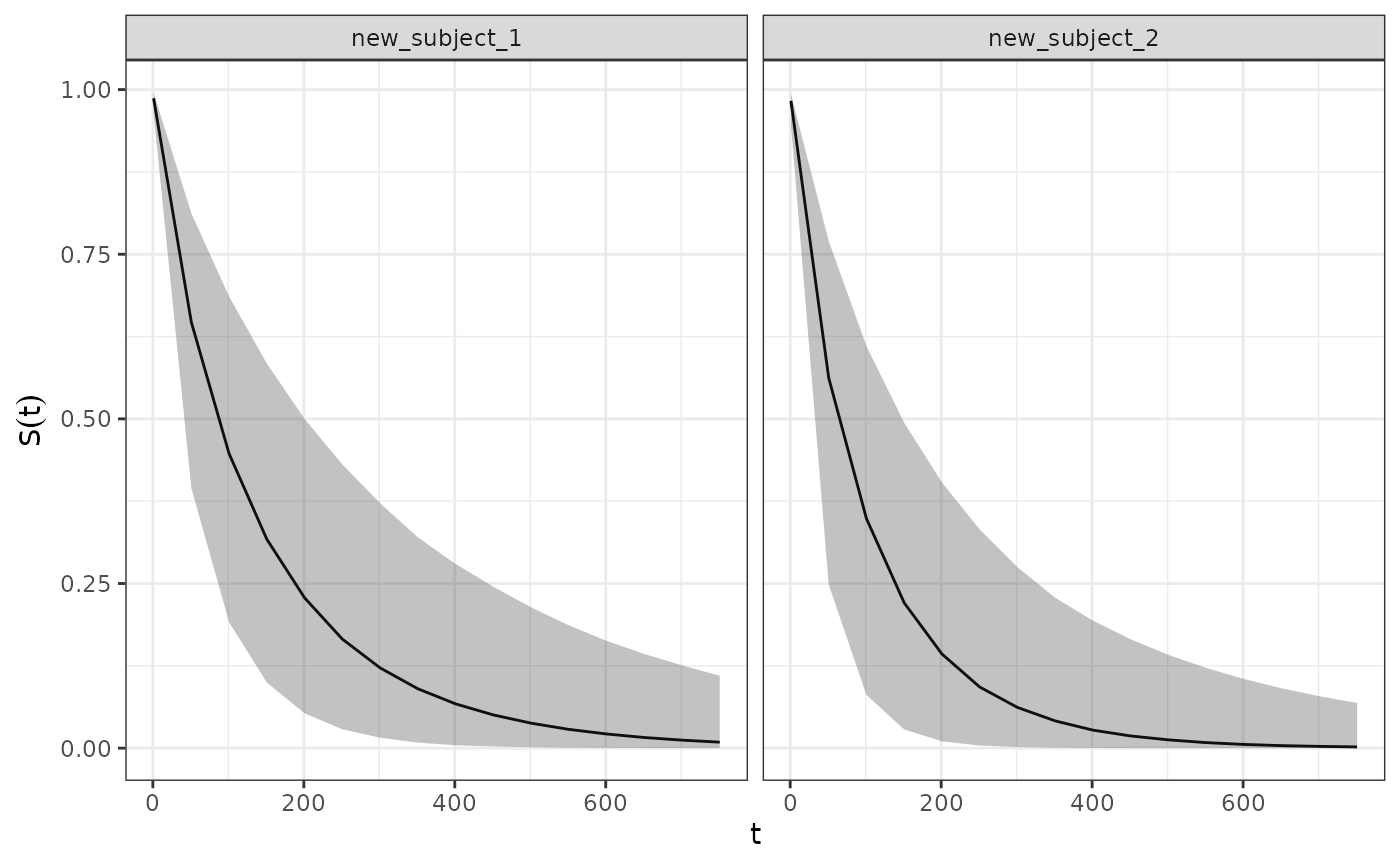
The exact names that are required for the params
argument (i.e. the longitudinal model parameters) can be found by
running getPredictionNames() on the longitudinal model
object. For example:
getPredictionNames(LongitudinalGSF())
#> [1] "b" "s" "g" "phi"Brier Score
The brierScore() method can be used to extract the Brier
Scores (predictive performance measure) from our
SurvivalQuantities object.
sq <- SurvivalQuantities(
mcmc_results,
grid = GridFixed(times = c(1, 50, 100, 400, 800)),
type = "surv"
)
brierScore(sq)
#> 1 50 100 400 800
#> 0.01951390 0.17167799 0.23677714 0.14894734 0.06115057Initial Values
By default jmpost will set the initial values for all
parameters to be a random value drawn from the prior distribution that
has been shrunk towards the mean of said distribution e.g. for a
prior_normal(4, 2) the initial value for each chain will
be:
4 * shrinkage_factor + rnorm(1, 4, 2) * (1 - shrinkage_factor)Note that the shrinkage factor is set to 0.5 by default and can be
changed via the jmpost.prior_shrinkage option e.g.
options("jmpost.prior_shrinkage" = 0.7)If you wish to manually specify the initial values you can do so via
the initialValues() function. For example:
joint_model <- JointModel(
longitudinal = LongitudinalRandomSlope(),
survival = SurvivalExponential(),
link = linkNone()
)
initial_values <- initialValues(joint_model, n_chains = 2)
initial_values[[1]]$lm_rs_intercept <- 0.2
initial_values[[2]]$lm_rs_intercept <- 0.3
mcmc_results <- sampleStanModel(
joint_model,
data = DataJoint(...),
init = initial_values,
chains = 2
)Note the following: - initialValues() will return a list
of lists where each sublist contains the initial values for the
corresponding chain index. - initialValues() by default
just returns 1 initial value for each parameter; the
sampleStanModel() function will then broadcast this number
as many times as required e.g. if you have 3 covariates in your survival
model then the initial value for the Beta coeficient will be repeated 3
times. If however you want to specify individual initial values for each
covariate you can do so by passing in a vector of the same length as the
number of covariates. - initialValues() will not check if
the proposed values are valid for constrained parameters. That is, if
you are using a prior_cauchy(0, 1) prior for a parameter
that should be >0 then you will need to manually set the
initial value (as described above) to ensure that it is a valid value. -
For constrained parameters (e.g. variance parameters that must be \(> 0\)) initialValues() will
continuously sample and discard initial values until it generates one
that meet the constraints. If after 100 attempts no valid initial value
has been found then it will throw an error.