Model-guided design of regulatory elements#
In this example, we use gReLU to perform model-guided design of accessible DNA elements for microglial cells.
import anndata
import numpy as np
import pandas as pd
import os
import importlib
import re
%matplotlib inline
Load the pre-trained Catlas model from the model zoo#
This is a binary classification model trained on snATAC-seq data from Catlas (http://catlas.org/humanenhancer/).
import grelu.resources
model = grelu.resources.load_model(repo_id="Genentech/human-atac-catlas-model", filename="model.ckpt")
View the model’s metadata#
model.data_params is a dictionary containing metadata about the data used to train the model. Let’s look at what information is stored:
model.data_params.keys()
dict_keys(['tasks', 'train', 'val', 'test'])
for k, v in model.data_params['train'].items():
if k !='intervals':
print(k, v)
bin_size 1
end both
genome hg38
label_aggfunc None
label_len 200
label_transform_func None
max_label_clip None
max_pair_shift 0
max_seq_shift 2
min_label_clip None
n_alleles 1
n_augmented 1
n_seqs 977014
n_tasks 204
padded_label_len 200
padded_seq_len 204
predict False
rc True
seq_len 200
Note the parameter seq_len. This tells us that the model was trained on 200 bp long sequences.
model.data_params['tasks'] is a large dictionary containing metadata about the output tracks that the model predicts. We can collect these into a dataframe called tasks:
tasks = pd.DataFrame(model.data_params["tasks"])
tasks.head(3)
| name | cell type | |
|---|---|---|
| 0 | Follicular | Follicular |
| 1 | Fibro General | Fibro General |
| 2 | Acinar | Acinar |
model.data_params['train']['intervals'] is a dictionary containing the genomic intervals used to train the model. Similarly, model.data_params['test']['intervals'] contains genomic intervals in the test set. We can collect these into a dataframe called test_intervals:
test_intervals = pd.DataFrame(model.data_params['test']['intervals'])
test_intervals.head(3)
| chrom | start | end | cre_class | in_fetal | in_adult | cre_module | width | cre_idx | enformer_split | split | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1 | 143497510 | 143497710 | Promoter Proximal | no | yes | 113 | 400 | 53530 | test | test |
| 1 | chr1 | 143498052 | 143498252 | Promoter Proximal | no | yes | 4 | 400 | 53531 | test | test |
| 2 | chr1 | 143498633 | 143498833 | Promoter | yes | no | 46 | 400 | 53532 | test | test |
Example 1: Starting from a random sequence, evolve a sequence with high accessibility in microglia using directed evolution.#
Generate a random starting sequence#
import grelu.sequence.utils
start_seq = grelu.sequence.utils.generate_random_sequences(
n=1, # number of sequences to generate
seq_len=model.data_params['train']['seq_len'], # Length of the generated sequence
seed=0,
output_format="strings"
)[0]
start_seq
'ATCATTTTCTCGATGAAAGCGTTGACCCCACATATCGTTAGTACTCTTGTACCCTATGATTGTGTAGAAACCGAACTACGGTACCTCCTGTTGGTAGTCACGATAGATTATAAAAGTATGTTCCCACCCTATCGACGAGACTGGCATCCTAGGTGTTTGCGGTGTTGGTACGTGCGCAGGTATGTAAGAGTGGTAAACGA'
Create the objective to optimize - predictions in microglia#
In the first example, we want to create a sequence that has a high probability of accessibility in microglia (regardless of what it does in other cell types). We first must define the objective that we want to optimize. Such objective functions are defined in grelu.transforms.prediction_transforms. We pick the Aggregate class, which tells the model to aggregate predictions over a subset of output tasks or positions.
from grelu.transforms.prediction_transforms import Aggregate
microglia_score = Aggregate(
tasks = ["Microglia"],
model = model,
)
Run directed evolution to maximize the prediction in microglia, ignoring other cell types#
grelu.design contains algorithms for model-guided sequence design. We pick the evolve function, which performs directed evolution. Note that it selects the sequence with the highest objective function value at every iteration, which in this case is the highest predicted accessibility in microglia. If you wanted to instead select the sequence with the lowest prediction in microglia, you could add weight=-1 to the Aggregate object.
import grelu.design
output = grelu.design.evolve(
[start_seq], # The initial sequences
model,
prediction_transform=microglia_score, # Objective to optimize
max_iter=5, # Number of iterations for directed evolution
num_workers=8,
devices=0,
return_seqs="all", # Return all the evolved sequences
)
Iteration 0
Best value at iteration 0: 0.241
Iteration 1
Best value at iteration 1: 0.352
Iteration 2
Best value at iteration 2: 0.621
Iteration 3
Best value at iteration 3: 0.820
Iteration 4
Best value at iteration 4: 0.912
Iteration 5
Best value at iteration 5: 0.957
Examine the output#
The output of grelu.design.evolve is a dataframe which contains all the sequences generated during directed evolution, and the model’s predictions. It also contains the position that was mutated and the base that was substituted at that position.
output.head()
| iter | start_seq | best_in_iter | prediction_score | seq_score | total_score | seq | position | allele | Microglia | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | True | 0.240814 | 0 | 0.240814 | ATCATTTTCTCGATGAAAGCGTTGACCCCACATATCGTTAGTACTC... | NaN | NaN | 0.240823 |
| 1 | 1 | 0 | False | 0.234945 | 0 | 0.234945 | CTCATTTTCTCGATGAAAGCGTTGACCCCACATATCGTTAGTACTC... | 0.0 | C | 0.234945 |
| 2 | 1 | 0 | False | 0.237114 | 0 | 0.237114 | GTCATTTTCTCGATGAAAGCGTTGACCCCACATATCGTTAGTACTC... | 0.0 | G | 0.237114 |
| 3 | 1 | 0 | False | 0.240810 | 0 | 0.240810 | TTCATTTTCTCGATGAAAGCGTTGACCCCACATATCGTTAGTACTC... | 0.0 | T | 0.240810 |
| 4 | 1 | 0 | False | 0.236174 | 0 | 0.236174 | AACATTTTCTCGATGAAAGCGTTGACCCCACATATCGTTAGTACTC... | 1.0 | A | 0.236174 |
We can visualize the model’s predictions on the sequences generated at each iteration:
import grelu.visualize
grelu.visualize.plot_evolution(output, outlier_size=.1)
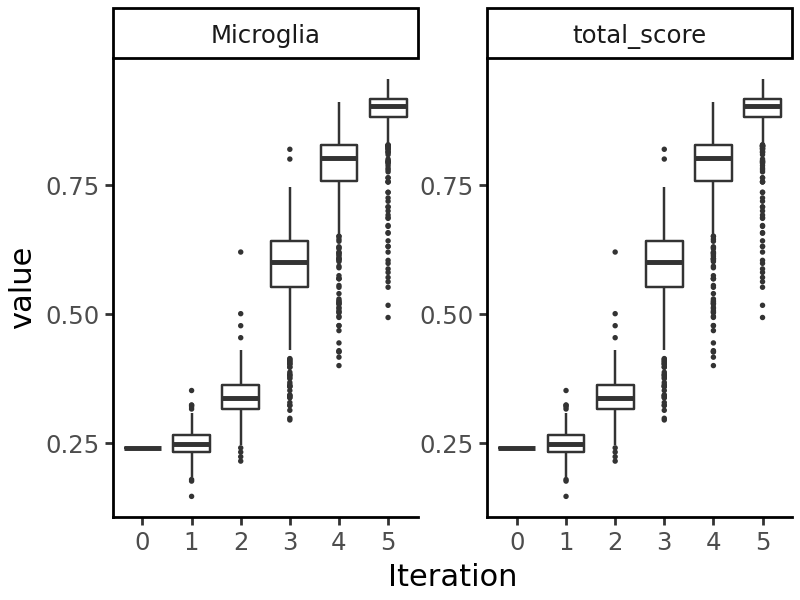
We see that the predicted probability of accessibility in Microglia increases at each iteration.
Compare the initial and final sequence#
Let us take the best sequence from the final iteration (iteration 5):
end_seq = output[output.iter==5].sort_values('total_score').iloc[-1].seq
end_seq
'ATCATTTTCTCGATGAAAGCGTTGACCCCACCTATCGTTAGTACTCTTGTACCCTATGATTGGGTAGAAACCGAACTACGGTACTTCCTGTTGGTAGTCACGATAGATTATAAAAGTATGTTCCCGCCCTATCGACGAGACTGGCATCCTAGGTGTTTGCGGTGTTGGTACGTGCGCAGGTCTGTAAGAGTGGTAAACGA'
We can compare this to the sequence we started with to see what changes were made:
import grelu.sequence.mutate
mutated_positions = grelu.sequence.mutate.seq_differences(start_seq, end_seq, verbose=True)
Position: 31 Reference base: A Alternate base: C Reference sequence: CCCACATATC
Position: 62 Reference base: T Alternate base: G Reference sequence: GATTGTGTAG
Position: 84 Reference base: C Alternate base: T Reference sequence: GGTACCTCCT
Position: 125 Reference base: A Alternate base: G Reference sequence: TTCCCACCCT
Position: 181 Reference base: A Alternate base: C Reference sequence: CAGGTATGTA
Next, we will use the grelu.interpret module to understand why these changes were made. One way to do this is by calculating per-base importance scores for the start and end sequence. gReLU provides several methods to do this, including In Silico Mutagenesis (ISM), DeepShap, InputxGradient and Integrated Gradients. Let’s first try inputxgradient on the start and end sequences.
import grelu.interpret.score
start_attrs = grelu.interpret.score.get_attributions(
model, start_seq, prediction_transform=microglia_score, device=0,
method="inputxgradient",
)
end_attrs = grelu.interpret.score.get_attributions(
model, end_seq, prediction_transform=microglia_score, device=0,
method="inputxgradient",
)
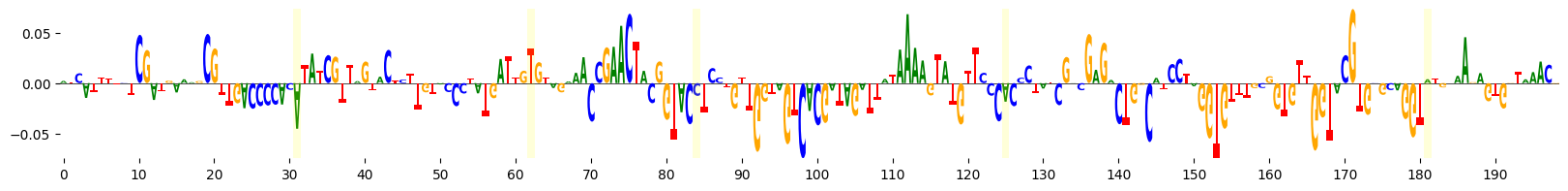
grelu.visualize.plot_attributions(
start_attrs,
highlight_positions=mutated_positions, # Highlight the mutated positions
ticks=10,
)
<logomaker.src.Logo.Logo at 0x1516777c7c70>
grelu.visualize.plot_attributions(
end_attrs,
highlight_positions=mutated_positions, # Highlight the mutated positions,
ticks=10,
)
<logomaker.src.Logo.Logo at 0x1516774bc580>
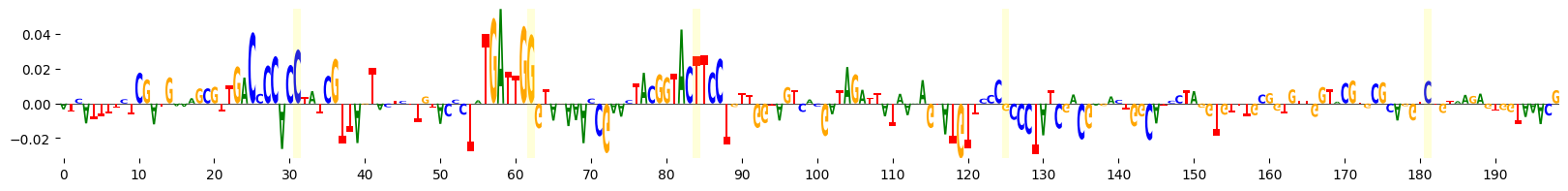
We see that several of the mutated bases are in regions of negative attributions, i.e. which reduce the model’s predictions.
Example 2: Use sequence constraints#
gReLU’s evolution function allows you to impose additional constraints upon the evolutionary process. For example, we can try to promote or avoid instances of a specific pattern or motif in our designed sequences. In this example, we suppose that we want to avoid occurrences of the sequence pattern CG or its reverse complement GC.
Let us count how many times this pattern occurs in our initial and final sequences.
unwanted_seqs = ["CG", "GC"]
n_initial = np.sum([len(re.findall(x, start_seq)) for x in unwanted_seqs])
n_final = np.sum([len(re.findall(x, end_seq)) for x in unwanted_seqs])
n_initial, n_final
(np.int64(17), np.int64(19))
Create additional design objective: A sequence transform#
We see that although directed evolution improved the predicted accessibility of the sequence in microglia, it also increased the number of unwanted sequence patterns. So, we want to add a penalty for these patterns in our evolutionary process. We accomplish this by creating another transform, this time one that operates on the sequence instead of the model’s predictions. These transforms are found in the grelu.transforms.seq_transforms module.
Here, we use the PatternScore class, which counts the number of times given subsequences occur in a sequence, and assigns the sequence a score based on this count. If you want to instead count the number of matches of a motif, use the MotifScore class.
from grelu.transforms.seq_transforms import PatternScore
We create an instance of this class that counts the occurrences of the unwanted subsequences and assigns a score of -0.1 for each occurrence. The negative sign means that sequences containing these subsequences will be penalized during evolution.
gc_penalizer = PatternScore(
patterns = unwanted_seqs, # Patterns to count
weights=[-.1, -.1], # Score to assign to each occurrence of each pattern
)
Run directed evolution including the sequence penalty#
output = grelu.design.evolve(
[start_seq], # The initial sequences
model,
prediction_transform=microglia_score, # Prediction objective to optimize
seq_transform=gc_penalizer, # Sequence objective to optimize
max_iter=5, # Number of iterations for directed evolution
num_workers=8,
devices=0,
return_seqs="all", # Return all the evolved sequences
)
Iteration 0
Best value at iteration 0: -1.459
Iteration 1
Best value at iteration 1: -1.237
Iteration 2
Best value at iteration 2: -1.048
Iteration 3
Best value at iteration 3: -0.878
Iteration 4
Best value at iteration 4: -0.749
Iteration 5
Best value at iteration 5: -0.638
Examine results#
Let’s examine the new optimized sequence and see whether it has fewer instances of GC and CG.
end_seq = output[output.iter==5].sort_values('total_score').iloc[-1].seq
np.sum([len(re.findall(x, end_seq)) for x in unwanted_seqs])
np.int64(9)
We see that the number of GC/CG occurrences has been reduced. When we visualize the output of evolution, we see that both the prediction objective (accessibility in microglia) and the sequence objective have been optimized:
grelu.visualize.plot_evolution(output, outlier_size=.1)
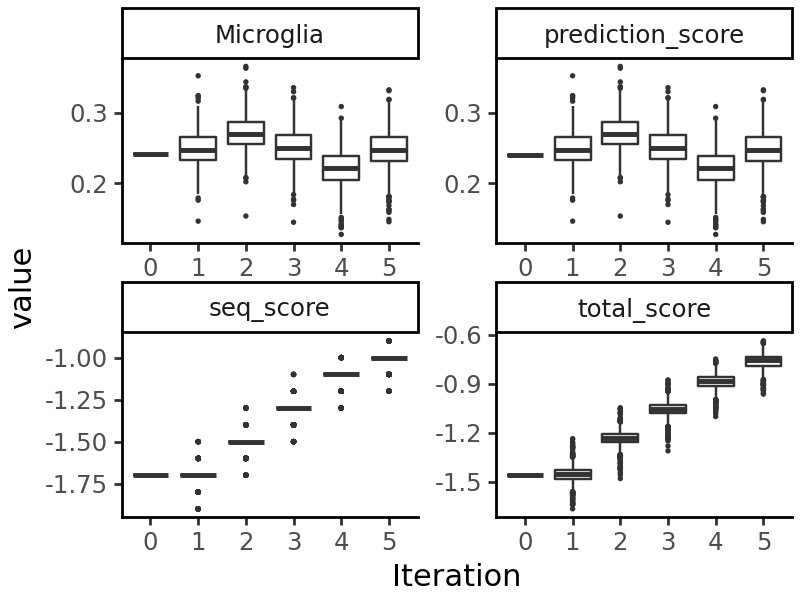
Example 3: Evolve a real genomic sequence toward microglia-specific accessibility using directed evolution#
In this example, we want to evolve a sequence that has a high predicted probability of being accessible in microglia, but low predicted probability of accessibility in other cell types (e.g. neurons). To increase the realism of the final sequence, we also want to start with a set of real genomic sequences instead of random sequences. These should be sequences that were not seen by the model during training or validation.
We can examine the model’s data_params to see which chromosomes were used for training and validation:
Select genomic starting sequences#
We now select some random genomic regions from the model’s test set:
start_intervals = test_intervals.sample(10, random_state=0)
start_intervals
| chrom | start | end | cre_class | in_fetal | in_adult | cre_module | width | cre_idx | enformer_split | split | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 14377 | chr14 | 29731170 | 29731370 | Distal | no | yes | 122 | 400 | 307182 | test | test |
| 714 | chr10 | 37865983 | 37866183 | Distal | yes | yes | 14 | 400 | 115934 | test | test |
| 20226 | chr14 | 49376887 | 49377087 | Distal | no | yes | 122 | 400 | 313045 | test | test |
| 63957 | chr3 | 183215891 | 183216091 | Distal | yes | yes | 34 | 400 | 733910 | test | test |
| 49631 | chr19 | 10618186 | 10618386 | Distal | yes | yes | 63 | 400 | 482008 | test | test |
| 47836 | chr15 | 25797712 | 25797912 | Distal | yes | yes | 143 | 400 | 341079 | test | test |
| 54616 | chr19 | 38034528 | 38034728 | Distal | yes | yes | 15 | 400 | 491075 | test | test |
| 63385 | chr22 | 22030660 | 22030860 | Promoter | yes | yes | 13 | 400 | 644921 | test | test |
| 41067 | chr14 | 95312376 | 95312576 | Distal | yes | no | 24 | 400 | 333904 | test | test |
| 68642 | chr3 | 192483010 | 192483210 | Distal | no | yes | 30 | 400 | 738595 | test | test |
Define design objective: cell type-specific accessibility in microglia#
We now want to optimize a more complicated objective. We want to create sequences that have a high probability of accessibility in microglia but low probability of accessibility in neurons. We begin by identifying neuron-related cell types in the model’s output:
neuron_tasks = tasks.name[tasks.name.str.contains("Neuron")]
print(neuron_tasks)
51 Enteric Neuron
109 Fetal Excitatory Neuron 3
110 Fetal Excitatory Neuron 4
111 Fetal Excitatory Neuron 5
112 Fetal Excitatory Neuron 6
113 Fetal Excitatory Neuron 7
114 Fetal Excitatory Neuron 8
125 Fetal Adrenal Neuron
145 Fetal Retinal Neuron
146 Fetal Enteric Neuron
148 Fetal Placental Neuron
164 Fetal Inhibitory Neuron 1
165 Fetal Inhibitory Neuron 2
166 Fetal Excitatory Neuron 9
167 Fetal Excitatory Neuron 10
202 Fetal Excitatory Neuron 1
203 Fetal Excitatory Neuron 2
Name: name, dtype: object
We now define our objective using the Specificity class of transforms. This class takes the model’s predictions and calculates a score by comparing predictions in defined on-target tasks (in this case Microglia) and off-target tasks (in this case, neurons). We can also specify a relative weight for the off-target tasks, how to aggregate the on- and off-target predictions, and soft thresholds for off-target expression.
from grelu.transforms.prediction_transforms import Specificity
mcg_specificity = Specificity(
on_tasks = "Microglia", # We want high predictions in these
off_tasks = neuron_tasks, # We want low predictions in these
on_aggfunc = "mean",
off_aggfunc = "max", # Compare the on-target prediction to the highest prediction in any off-target task.
model=model,
compare_func="subtract", # Each sequence will get a score which is the ratio of the on-target and off-target scores
)
Run directed evolution#
Note that here, we start with multiple sequences and set for_each=False. This means that the model will evaluate all the starting sequences, choose the best one (with highest microglia-specific predicted accessibility) and continue with that sequence. If we set for_each=True instead, we would run directed evolution independently on each starting sequence. That option can be used to generate a more diverse set of evolved sequences.
output = grelu.design.evolve(
start_intervals, # Start from the natural sequences
model,
genome="hg38",
prediction_transform=mcg_specificity, # Optimize the specific accessibility score
max_iter=5,
num_workers=8,
devices=0,
return_seqs="all",
for_each=False,
)
Iteration 0
Best value at iteration 0: 0.284
Iteration 1
Best value at iteration 1: 0.808
Iteration 2
Best value at iteration 2: 0.918
Iteration 3
Best value at iteration 3: 0.956
Iteration 4
Best value at iteration 4: 0.967
Iteration 5
Best value at iteration 5: 0.977
Visualize the output#
We can plot the accessibility of the sequences at each iteration:
grelu.visualize.plot_evolution(output, figsize=(6, 14), outlier_size=0.2)
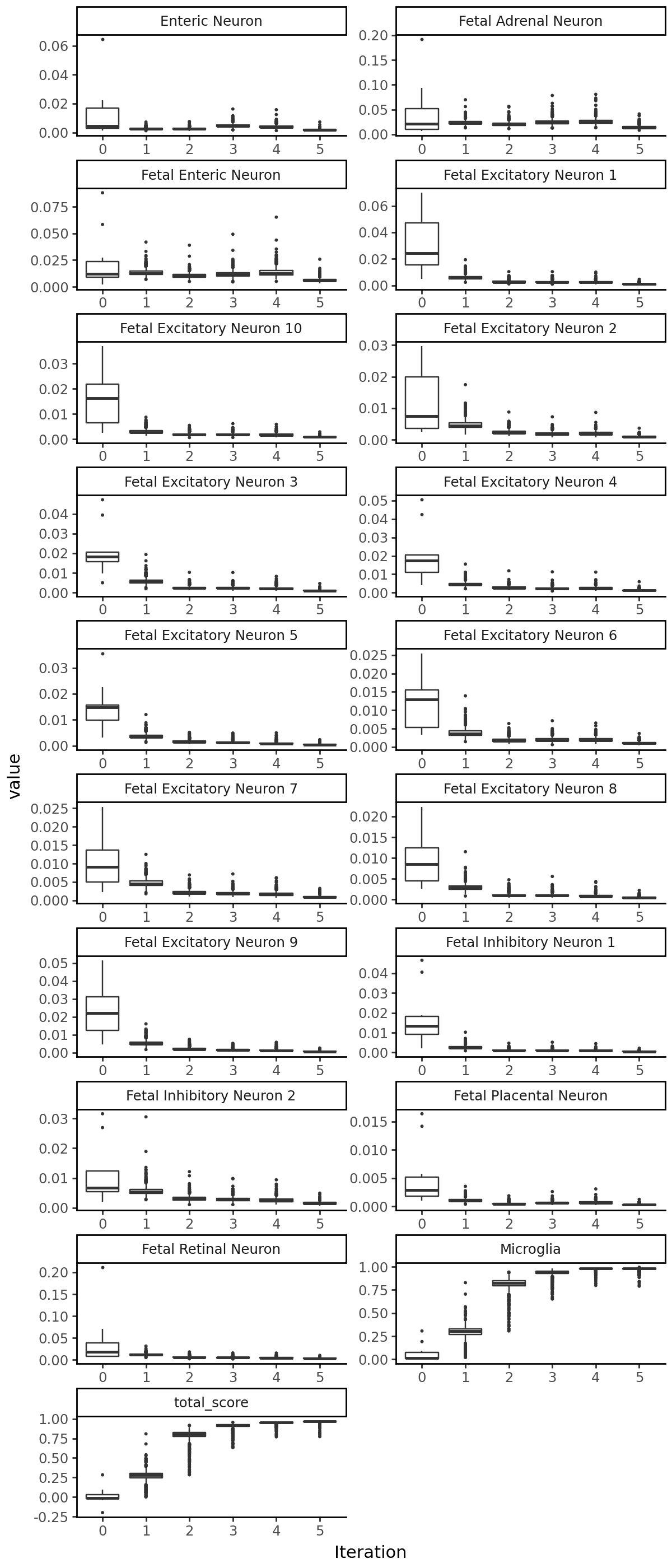
Compare start and end sequences#
start_seq = output[output.iter==0].sort_values('total_score').iloc[-1].seq
end_seq = output[output.iter==5].sort_values('total_score').iloc[-1].seq
mutated_positions = grelu.sequence.mutate.seq_differences(start_seq, end_seq, verbose=True)
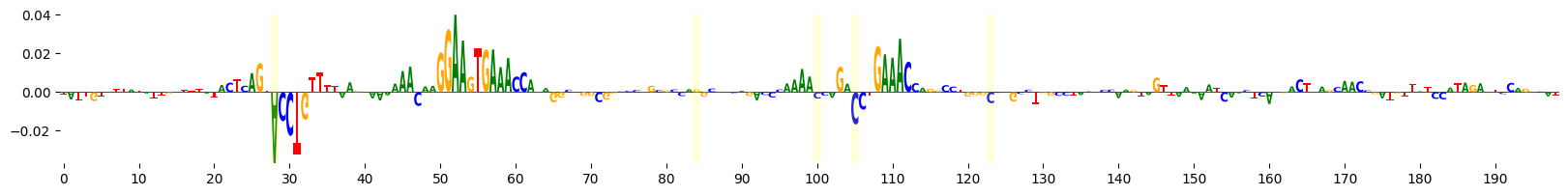
Position: 28 Reference base: A Alternate base: T Reference sequence: TCAGTACCTG
Position: 84 Reference base: G Alternate base: T Reference sequence: CGCCAGGCAC
Position: 100 Reference base: C Alternate base: G Reference sequence: CAAAACCAGA
Position: 105 Reference base: C Alternate base: A Reference sequence: CCAGACCTGA
Position: 123 Reference base: C Alternate base: G Reference sequence: CTGGGCCTGC
This time, let’s use Integrated Gradients to visualize the importance of these bases that were mutated at each iteration of evolution. Note also that we supply prediction_transform=mcg_specificity, so that we calculate the importance of each base to the specificity of accessibility in microglia.
start_attrs = grelu.interpret.score.get_attributions(
model, start_seq, prediction_transform=mcg_specificity, device=0, method="integratedgradients",
)
end_attrs = grelu.interpret.score.get_attributions(
model, end_seq, prediction_transform=mcg_specificity, device=0, method="integratedgradients",
)
grelu.visualize.plot_attributions(start_attrs, highlight_positions=mutated_positions)
<logomaker.src.Logo.Logo at 0x15166cb26620>
grelu.visualize.plot_attributions(end_attrs, highlight_positions=mutated_positions)
<logomaker.src.Logo.Logo at 0x15166ce9b0d0>
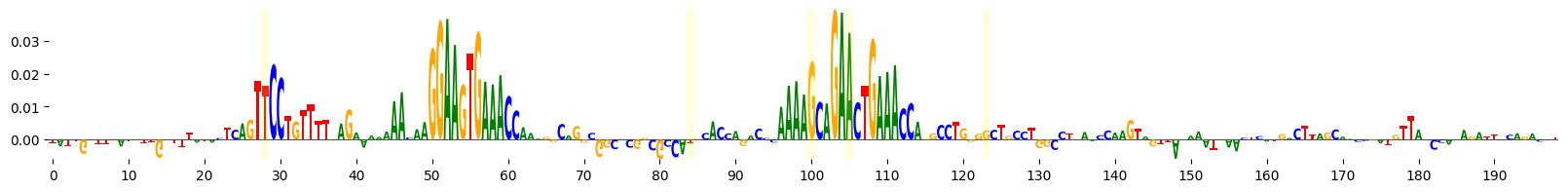
It seems that the evolution process has created new motifs. We can identify the specific motifs that were altered by scanning the reference and alternate sequence with HOCOMOCO motifs and comparing the output.
import grelu.interpret.motifs
comparison = grelu.interpret.motifs.compare_motifs(
ref_seq = start_seq,
alt_seq = end_seq,
motifs="hocomoco_v13",
pthresh=1e-4,
rc=True, # Scan both strands of the sequence
ref_attrs = start_attrs, # Attributions are optional
alt_attrs = end_attrs,
)
comparison
| motif | start | end | strand | fimo_p-value_alt | fimo_p-value_ref | fimo_score_alt | fimo_score_ref | motif_attr_score_alt | motif_attr_score_ref | site_attr_score_alt | site_attr_score_ref | fimo_score_diff | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ZN778.H13CORE.1.P.B | 60 | 91 | - | 0.964214 | 0.000080 | -87.077140 | -7.727288 | 0.000213 | 0.000128 | 0.000148 | 0.000125 | -79.349852 |
| 1 | ZN416.H13CORE.0.P.C | 118 | 141 | - | 0.926214 | 0.000055 | -36.351458 | 9.961223 | 0.000223 | -0.000516 | 0.000425 | -0.000318 | -46.312681 |
| 2 | P63.H13CORE.0.PS.A | 111 | 130 | - | 0.358806 | 0.000014 | -28.462253 | 13.368946 | 0.001106 | 0.000240 | 0.001184 | 0.000493 | -41.831200 |
| 3 | HAND2.H13CORE.1.P.B | 95 | 109 | - | 0.761428 | 0.000020 | -26.859183 | 12.592777 | 0.004862 | 0.000588 | 0.004870 | 0.000674 | -39.451960 |
| 4 | ZN322.H13CORE.0.P.C | 69 | 89 | - | 0.858887 | 0.000077 | -25.160535 | 10.122780 | -0.000170 | 0.000043 | -0.000258 | -0.000062 | -35.283315 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 61 | EHF.H13CORE.0.P.B | 22 | 37 | - | 0.000087 | 0.798337 | 10.568862 | -52.762712 | 0.006980 | 0.000368 | 0.002308 | -0.000934 | 63.331574 |
| 62 | ELF4.H13CORE.1.M.B | 23 | 38 | - | 0.000069 | 0.829283 | 10.363427 | -55.107857 | 0.007185 | 0.000401 | 0.002295 | -0.001065 | 65.471283 |
| 63 | SPIB.H13CORE.1.S.C | 24 | 37 | - | 0.000015 | 0.662778 | 11.185361 | -60.324991 | 0.008494 | 0.000243 | 0.002583 | -0.001306 | 71.510352 |
| 64 | ZN184.H13CORE.0.P.B | 96 | 127 | - | 0.000058 | 0.712160 | -8.513401 | -81.023293 | 0.005596 | -0.000245 | 0.003184 | 0.000946 | 72.509892 |
| 65 | SPI1.H13CORE.0.P.B | 23 | 37 | - | 0.000002 | 0.954894 | 15.881978 | -63.251506 | 0.007827 | -0.000573 | 0.002464 | -0.001088 | 79.133484 |
66 rows × 13 columns
comparison[comparison.motif=="SPI1.H13CORE.0.P.B"].sort_values("start")
| motif | start | end | strand | fimo_p-value_alt | fimo_p-value_ref | fimo_score_alt | fimo_score_ref | motif_attr_score_alt | motif_attr_score_ref | site_attr_score_alt | site_attr_score_ref | fimo_score_diff | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 65 | SPI1.H13CORE.0.P.B | 23 | 37 | - | 0.000002 | 0.954894 | 15.881978 | -63.251506 | 0.007827 | -0.000573 | 0.002464 | -0.001088 | 79.133484 |
| 48 | SPI1.H13CORE.0.P.B | 96 | 110 | + | 0.000029 | 0.005027 | 11.530125 | -5.399882 | 0.015494 | 0.004283 | 0.005231 | 0.000998 | 16.930007 |
The fimo_score_ref column contains the score for the motif in the original sequence and the fimo_score_alt column contains the score in the designed sequence. Similarly, the fimo_p-value_ref column contains the p-value for the motif in the original sequence and the fimo_p-value_alt column contains the p-value in the designed sequence.
site_attr_score is the average attribution value for all nucleotides within the motif match site. This score gives the importance of the sequence region but does not reflect the similarity between the PWM and the shape of the attributions.
motif_attr_score is the sum of the element-wise product of the PWM and the attributions. This score is higher when the shape of the motif matches the shape of the attribution profile, and is useful for ranking multiple motifs that all match to the same sequence region.
We see that the evolution process has created new instances of the SPI1.H13CORE.0.P.B motif.
Example 4: Evolution by motif scanning#
So far, we have been performing directed evolution by in silico mutagenesis: at each iteration, we create all possible single-base substitutions and choose the best one. Another approach is to insert a given motif into every possible position in the starting sequence, and choose the best one. We can run this approach by choosing method="pattern" in grelu.design.evolve.
Let’s start by getting the sequence of the SPI1.H12CORE.0.P.B motif.
import grelu.io.motifs
motifs = grelu.io.motifs.read_meme_file("hocomoco_v12", names=["SPI1.H12CORE.0.P.B"])
patterns = grelu.interpret.motifs.motifs_to_strings(motifs)
print(patterns)
['AAAAGAGGAAGTGA']
Run evolution#
We will now try to run the evolution function for microglia-specific accessibility, this time by scanning the starting sequence with the motif and finding the best position to substitute in this motif. You can do this with multiple motifs with different weights or penalties.
In addition, we demonstrate another constraint in the grelu.design.evolve function; you can constrain which positions can be altered, whether by ISM or by motif substitution. Here, we will specify that the motif should only be substituted in the first 100 bases of the sequence.
output = grelu.design.evolve(
start_intervals, # Start from the natural sequences
model,
genome="hg38",
prediction_transform=mcg_specificity, # Optimize the specific accessibility score
max_iter=2, # Since we are modifying a whole motif, we only run 2 iterations
method="pattern", # Evolution by pattern substitution
patterns=patterns, # SPIB motif
num_workers=8,
devices=0,
return_seqs="all",
for_each=False,
positions=range(100), # Positions allowed for substitution
)
Iteration 0
Best value at iteration 0: 0.284
Iteration 1
Best value at iteration 1: 0.907
Iteration 2
Best value at iteration 2: 0.951
grelu.visualize.plot_evolution(output, figsize=(5,10))
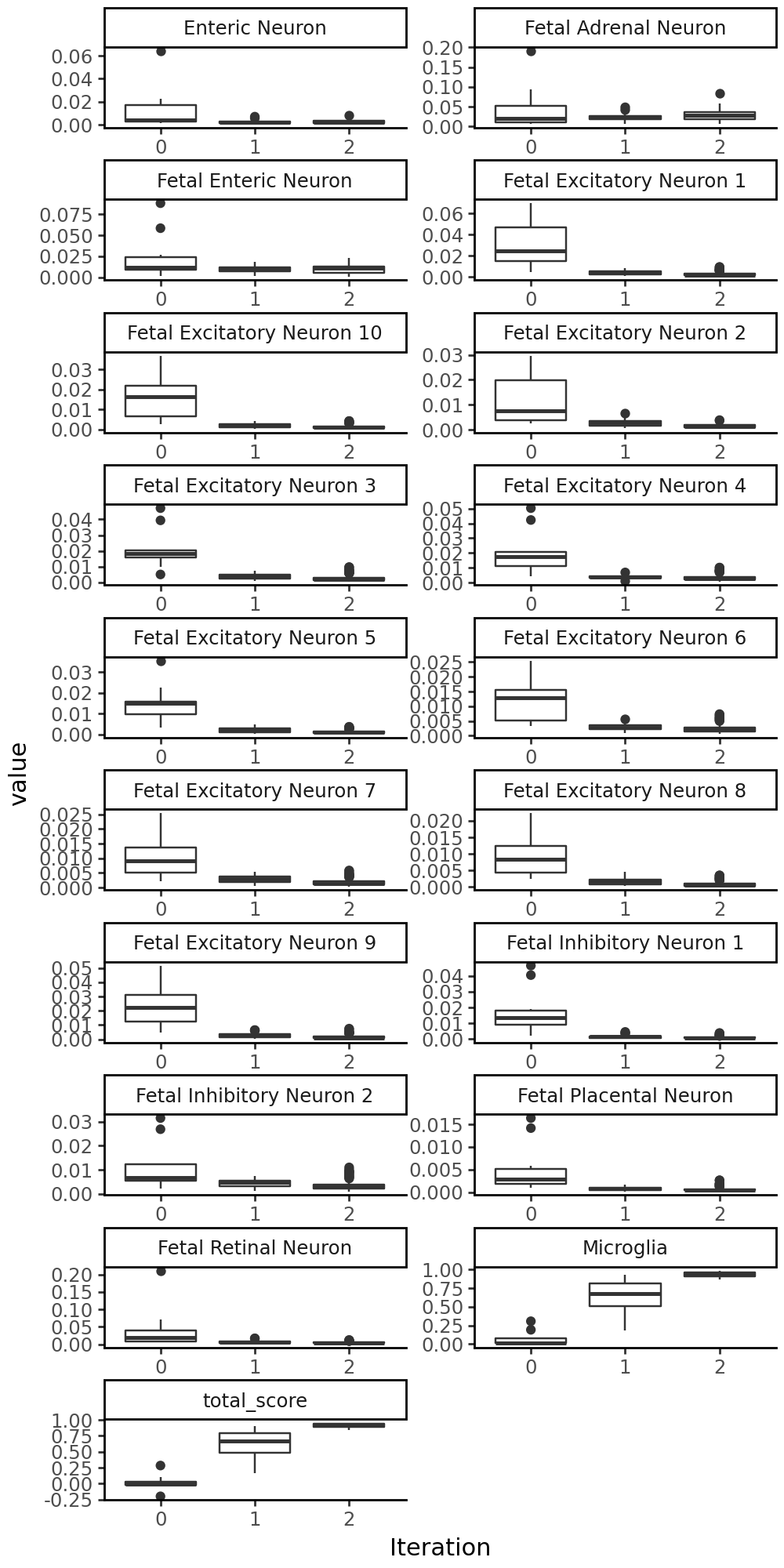
Let’s see where the motifs were inserted:
end_seq = output[output.iter==2].sort_values('total_score').iloc[-1].seq
end_attrs = grelu.interpret.score.get_attributions(
model, end_seq, prediction_transform=mcg_specificity, device=0, method="integratedgradients",
)
grelu.visualize.plot_attributions(end_attrs)
<logomaker.src.Logo.Logo at 0x151669e397b0>
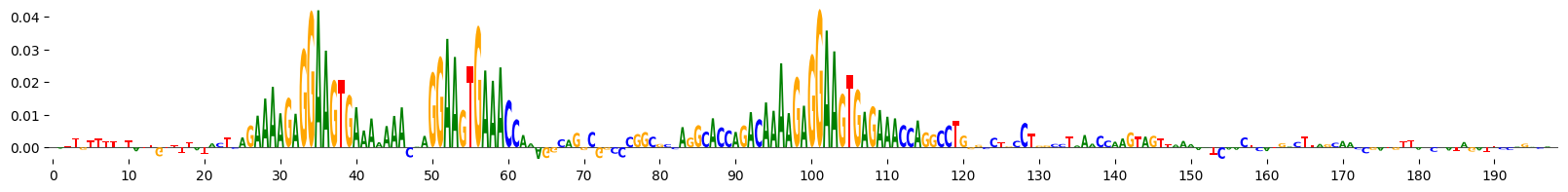
Example 5: Ledidi#
We also offer the Ledidi method by Schreiber et al. (2020) (https://www.biorxiv.org/content/10.1101/2020.05.21.109686v1). This is a gradient-based approach for sequence optimization. Here, we run Ledidi for the same objective as in example 3.
Ledidi requires a single starting sequence.
start_seq
'TATTGTTTTATATTGTTTTATACTCAGTACCTGTTTTAAGAAAAAAACAAGGAAGTGAAACCAAAGGCAGGCGGCCCGGCGCCAGGCACCAGACCCAAAACCAGACCTGAAACCAGGCCTGGGCCTGCCTGGCCTAAACCAAGTAGTTAAAAATCAACTCATGACTTAGCAACCGATGTTATCCATAGATTCCAGACATT'
end_seqs = grelu.design.ledidi(
start_seq,
model,
prediction_transform=mcg_specificity,
max_iter=2000,
devices=0,
lr=0.01,
l=0.01,
)
Running Ledidi
iter=I input_loss=0.0 output_loss=-0.2841 total_loss=-0.2841 time=0.0
iter=100 input_loss=0.1875 output_loss=-0.2844 total_loss=-0.2825 time=1.515
iter=200 input_loss= 1.0 output_loss=-0.3191 total_loss=-0.3091 time=1.381
iter=300 input_loss=9.125 output_loss=-0.8163 total_loss=-0.725 time=1.383
iter=400 input_loss=12.25 output_loss=-0.9189 total_loss=-0.7964 time=1.385
iter=500 input_loss=12.25 output_loss=-0.9339 total_loss=-0.8114 time=1.388
iter=600 input_loss=12.38 output_loss=-0.9435 total_loss=-0.8198 time=1.403
iter=F input_loss=10.88 output_loss=-0.955 total_loss=-0.8462 time=8.812
Cleaning up model state...
This returns a list of optimized sequences.
Let’s look at the first sequence in the list:
end_seqs[0]
'TATTGTTTTATATTGCTTTATACTCAGTACTTGTTTTAAGAAAAAAAGAAGGAAGTGAAACCAAAGGCAGGCGGCCCGGCGCCAGTCACCAGACCCAAAAGGAGAAGTGAAACCAGGGCTGGGCCTGCCTGGCCTAAACCAAGTAGTTAAAAATCCACTCATGACTTAGCAACCGATGTTATCCATAGATTCCAGACATT'
mutated_positions = grelu.sequence.mutate.seq_differences(start_seq, end_seqs[0])
Position: 15 Reference base: T Alternate base: C Reference sequence: TATTGTTTTA
Position: 30 Reference base: C Alternate base: T Reference sequence: AGTACCTGTT
Position: 47 Reference base: C Alternate base: G Reference sequence: AAAAACAAGG
Position: 85 Reference base: G Alternate base: T Reference sequence: GCCAGGCACC
Position: 100 Reference base: C Alternate base: G Reference sequence: CAAAACCAGA
Position: 101 Reference base: C Alternate base: G Reference sequence: AAAACCAGAC
Position: 105 Reference base: C Alternate base: A Reference sequence: CCAGACCTGA
Position: 106 Reference base: C Alternate base: G Reference sequence: CAGACCTGAA
Position: 117 Reference base: C Alternate base: G Reference sequence: CCAGGCCTGG
Position: 155 Reference base: A Alternate base: C Reference sequence: AAATCAACTC
We can compute the score for the original sequence and each final sequence:
mcg_specificity.compute(model.predict_on_seqs(start_seq)).squeeze()
array(0.28424257, dtype=float32)
mcg_specificity.compute(model.predict_on_seqs(end_seqs)).squeeze()
array([0.96382785, 0.9577518 , 0.94321746, 0.9384998 , 0.9697705 ,
0.966124 , 0.9532623 , 0.96199113, 0.9583562 , 0.93500376,
0.9560423 , 0.95118874, 0.9586166 , 0.9589285 , 0.95083815,
0.9561787 ], dtype=float32)